2020 年 1 月,整个中国因新型冠状病毒 2019-nCov 得了一场“感冒”。疫情一开始,武汉病毒研究所石正丽团队就用实验证实了血管紧张素转化酶 2(Angiotensin-converting enzyme 2,ACE2)是新型冠状病毒感染人体的受体基因。2020 年 1 月 26 日,上海同济大学医学院左为研究团队在《bioRxiv》上发表了题为“Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov”的文章。
该研究利用已有的数据库,结合单细胞 RNA 测序技术中相关生信分析,对 ACE2 在人肺内单个细胞的表达情况进行了分析,共涉及 8 个样本,43134 个细胞。结果表明:ACE2 受体主要在一部分(1% 左右)II 型肺泡上皮细胞(AT2) 中表达;同时发现这些 AT2 细胞除了表达病毒受体,还表达与病毒复制和传播相关的基因,说明其很可能是冠状病毒的靶细胞。可见,单细胞测序技术不仅是科研的利器,同时还为破解疫情做了应有的贡献。那么,1 月份利用单细胞测序技术在基础研究中还有哪些突破呢?
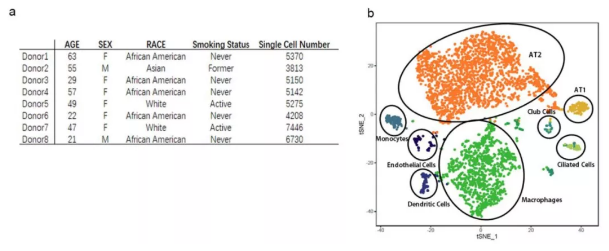
图 1 人肺的单细胞分析
1 月份搜集到高分文献(IF>10)26 篇,表 1 按照在线时间顺序列出了文章所在期刊、研究方向,分选平台以及英文题目等。
表 1 2020 年 1 月单细胞测序高分文献集锦 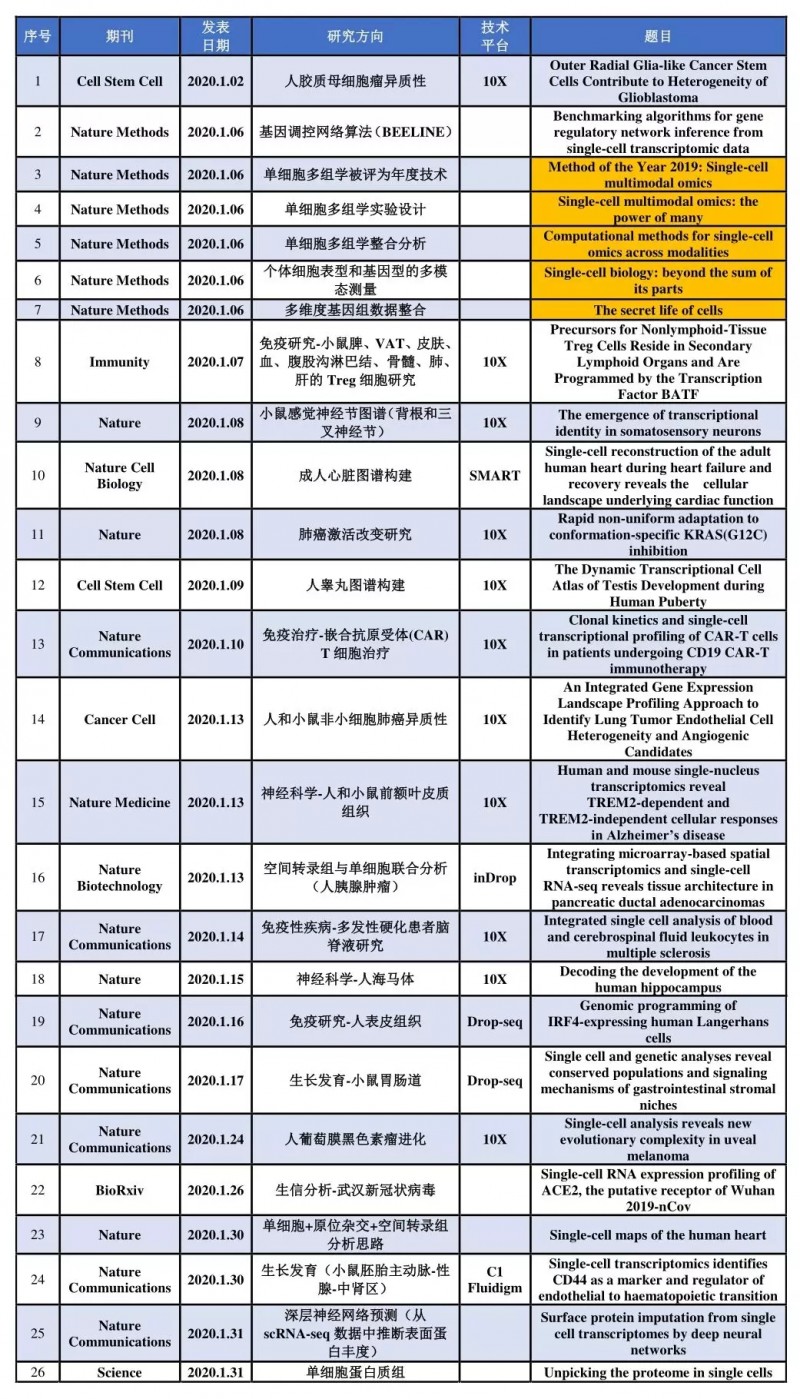
我们知道 2011 年,《Nature Methods》将单细胞测序列为当年度值得期待的技术之一;2013 年 1 月,《Science》杂志将单细胞测序列为年度值得关注的六大领域榜首;2014 年 1 月,《Nature Methods》将单细胞测序列为 2013 年度重要的方法学进展。让人兴奋的是 2020 年 1 月 6 号《Nature Methods》将单细胞多组学分析选为了“Method of the Year 2019”,并在线发表 5 篇相关文章(表 1 中标黄部分)。

其中,加利福尼亚大学圣地亚哥分校任兵教授在文章“Single-cell multimodal omics: the power of many”中分享了单细胞多组学的实验设计。文章首先总结现有的单细胞多组学测序技术的应用范围,包括细胞核内 DNA,RNA,DNA 甲基化,染色体开发程度,染色体三维结构,胞质 RNA,线粒体基因组,细胞表面蛋白质以及基于 CRISPR 的测序技术。
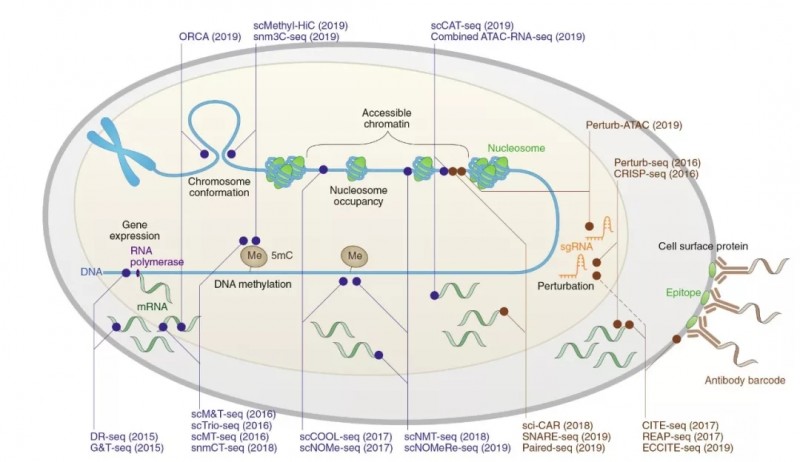
图 2 已开发的同时分析单细胞表观遗传特征、DNA 序列、基因表达变异和细胞表面蛋白的方法。蓝色代表低通量高成本的测序技术,棕色代表高通量低成本的测序技术。
然后作者总结现有技术的不足和发展的方向,比如单细胞基因检测数较少(可通过实验设计改善);单细胞蛋白组学不成熟(提高灵敏度和通量);空间测序技术只用于转录组(应扩展到其他组学)。
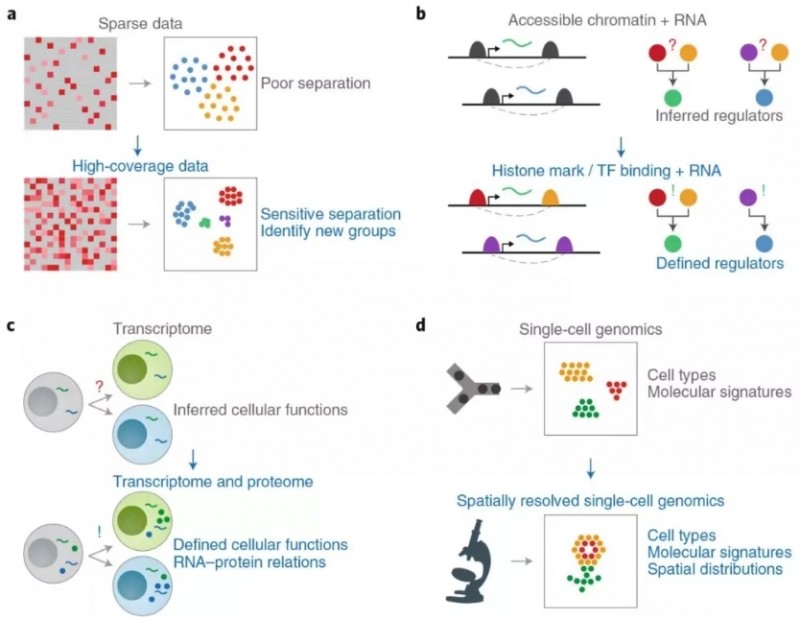
图 3 现有单细胞多组学技术的不足和发展方向
同时在线的另外一篇文章“Computational methods for single-cell omics across modalities”则讲述了单细胞多组学联合分析方法。分析对象:整合不同数据对象(ATAC,RNA,蛋白)的同时,保留细胞类型固有生物学差异。实际问题:批次效应处理,ATAC/RNA 整合,CITE-seq 数据分析等。基本方法:CCA/ 非负矩阵分解 / 自编码等。基本原理:多组学整合的思路与批次效应去除的思路相似,combat 是基于线性回归的批次效应去除方法,CCA 是 Seurat 刚开始处理批次效应的基本方法,后来用于 ATAC/RNA 整合的方法 anchor 也是基于 CCA,ATAC/RNA 整合的方法也可以用非负矩阵分解。未来可期:RNA 和 ATAC 组合可以促进调控网络研究;空间数据可以辅助细胞间相互作用的分析;整合 Bulk 数据和单细胞数据。
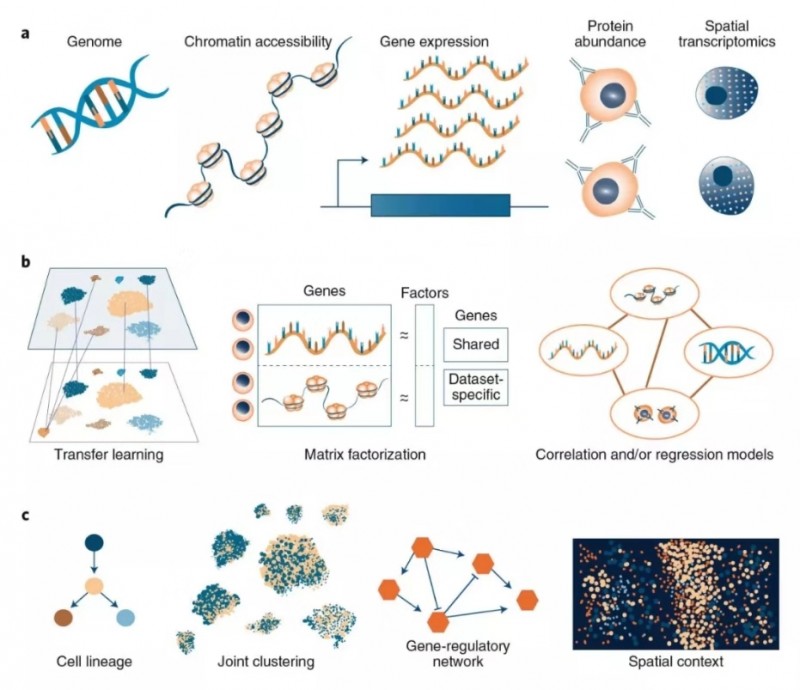
图 4 单细胞多组学联合分析不同计算方法的示意图
1 月份单细胞测序另一个突破就是和空间转录组实现强强联合,以获得目标组织的三维空间转录组图谱。2020 年 1 月 13 日,《Nature Biotechnology》上发表了“Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas”的文章。作者对 3 个胰腺导管癌(PDAC)组织 A、B、C 进行 scRNA-seq(indrop-seq),同时对 2 个主要样本(A、B,各 3 张切片),4 个附加样本(D、E、F 和 G,各 1 张切片),共 10 张切片做空间转录组(ST)。样品 A 和 B 的 scRNA-seq 和 ST 的数据集信息通过多模式相交分析(Multimodal intersection analysis,MIA)进行整合,就是用超几何算法检验细胞亚群的 marker 基因与组织亚区的 marker 基因重叠的数目是否达到显著水平,若显著,则认为该细胞亚群与该组织亚区相对应。
作者确定了组织中亚区中特定的细胞类型后,通过进一步细分细胞亚群和组织亚区,再次利用 MIA 算法进行映射,获得了更为详细的细胞位置 - 类型信息。作者利用得到的 MIA 结果绘制了不同肿瘤样本微环境的特点,免疫环境状态,应激水平以及细胞之间相互作用的模式,有助于预判患者预后。可见,MIA 算法的开发可将高分辨率的 scRNA-seq(单细胞水平)和带空间位置信息的 ST 结合,为全面解析组织生物学信息提供了技术支撑。
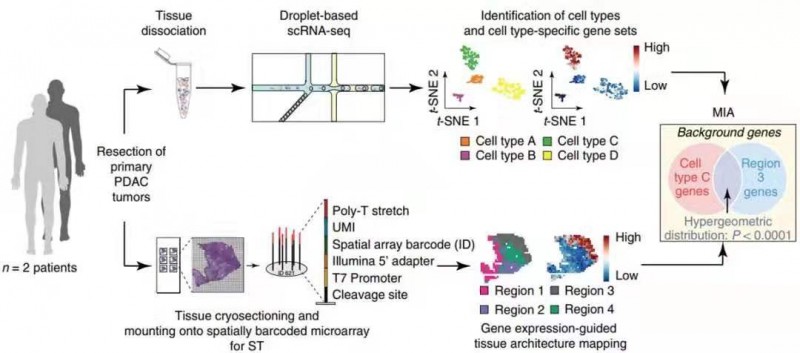
图 5 实验设计与分析示意图。手术切除的 PDAC 肿瘤同时进行 scRNA-seq 和 ST 处理。聚类后,根据特异表达的基因推断每个簇的细胞类型。剩余组织的冷冻切片用于 ST 分析,其中每个点捕获组织中特定位置细胞中的转录组。将多模式相交分析(MIA)应用于两个数据集,揭示了各种细胞类型的空间分布。
