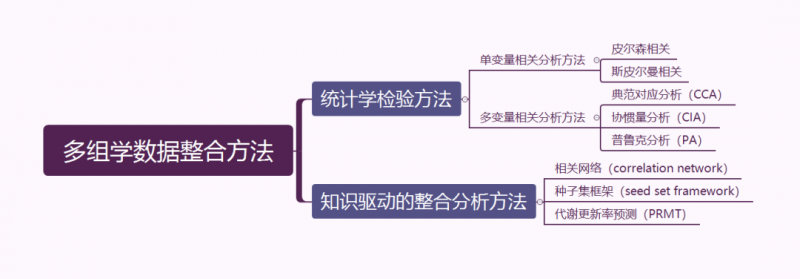
微生物组与代谢组的数据整合方法按照是否基于已有知识可以分为统计学检验方法和知识驱动的整合分析方法。统计学检验方法采用单变量或多变量分析阐述不同组学层级所属生物学指标之间的相关性。单变量相关分析分析过程相对简单,假阳性率高,需要多重校正检验以控制Ⅰ型错误率,因此并不适用于分析多组学数据。多变量相关分析可以考虑到复杂样本中多个指标的相关性,由于测序和质谱测量之间的测量单位不同,这种方法不太适用于微生物与代谢物的相互作用。知识驱动的整合分析方法是将单一组学层级获取的生物学指标投射到已有数据库中,以解释各生物学指标之间的相互联系,该方法包括常用的构建相关性网络、种子集框架和通过预测代谢更新率构建代谢模型,从而将微生物群落与代谢物相关联。
本片文章中将主要介绍伯豪现有的两种微生物组与代谢组联合分析方法——mmvec 和 MIMOSA 2。mmvec 主要考虑到微生物与代谢物的共现概率来预测可能的微生物与代谢物的相互作用;MIMOSA 2 对 PRMT 的功能进行了扩展,整合代谢组学数据研究微生物组成和代谢活性。这两种分析方法对目前常用的联合分析方法进行了进一步的补充,结果相对更加可信。
微生物与代谢组联合分析方法 —— mmvec 神经元网络介绍
2019 年 11 月 Rob Knight 团队在《Nature Methods》发表了一篇“Learning representations of microbe-metabolite interactions”提出 mmvec (microbe-metabolite vectors) 神经元网络,该方法通过学习代谢物和微生物共现概率(共现概率是指在观察到微生物的情况下观察代谢物的条件概率,从而使我们能够确定可能的微生物与代谢物的相互作用),对代谢物与微生物互作进行评级,并可视化显示分析结果。

mmvec 分析原理示意图
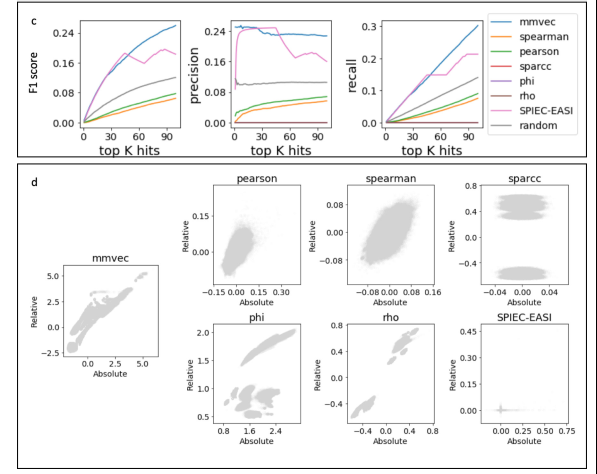
分析比较
文章中作者将 mmvec 与 Pearson,Spearman,SPIEC-EASI,SparCC 和 Proportionality 等分析方法进行了比较,证明 mmvec 的 F1 score、高精度和召回率更高,且该模型具有一定的鲁棒性。同时,研究人员以已知的环境(沙漠土壤湿润生物结壳)和临床(囊性纤维化肺)实例为例,展示了这一方法恢复微生物与代谢物之间关系的能力,并证明了该方法如何发现微生物产生的代谢产物与炎症性肠病之间的关系。
微生物与代谢组联合分析方法 —— MIMOSA 2 介绍
2016 年 Borenstein Lab 提出 MIMOSA 模型,该方法对 PRMT 进行了扩展,通过预测微生物群落代谢能力——CMP(community-wide metabolite potential)构建一个代谢模型来预测群落组成对代谢物浓度的影响,并评估该预测值是否与测量到的代谢组学特征相符,从而将物种组成和代谢物浓度相联系,该方法在 2018 年进行了升级——MIMOSA 2。
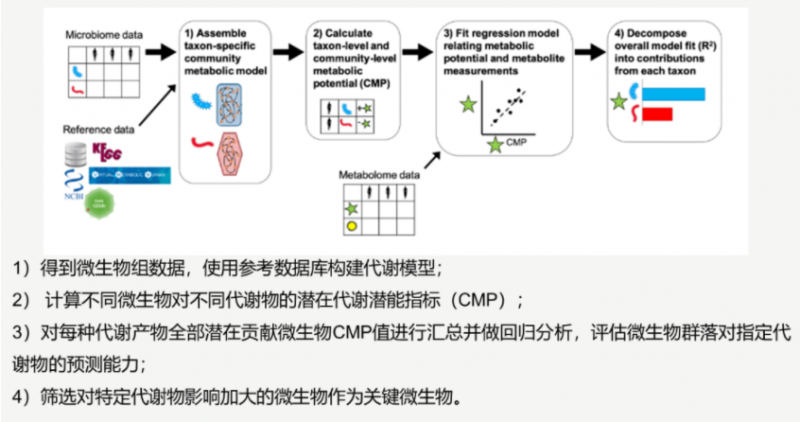
通过 MIMOSA 2 分析主要可以回答一下三个问题:
1)代谢组数据是否和微生物组数据紧密相关;
2)微生物群落差异是否可以解释代谢差异;
3)何种微生物或者基因造成了代谢差异,或者贡献了代谢差异。
结果展示
1. mmvec 神经元网络主要结果展示
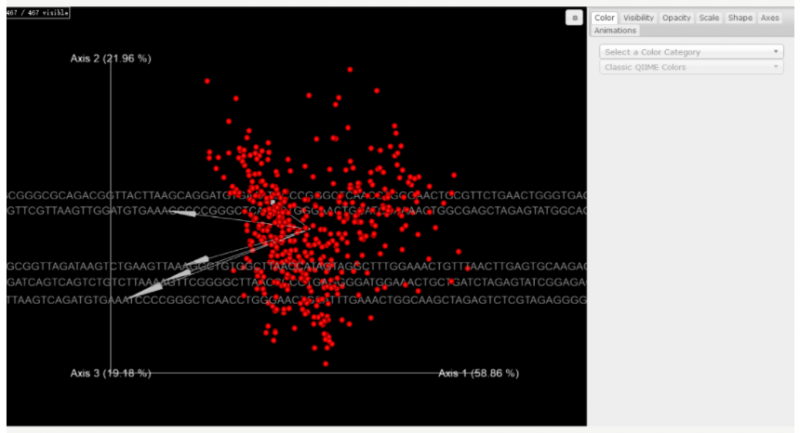
▲微生物和代谢物共现概率 biplot
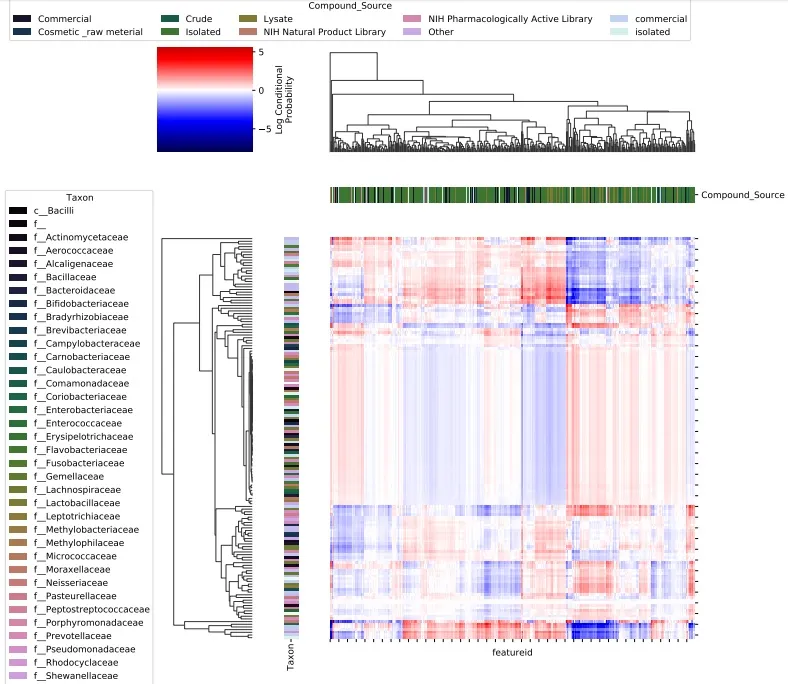
▲微生物和代谢物共现概率 heatmap
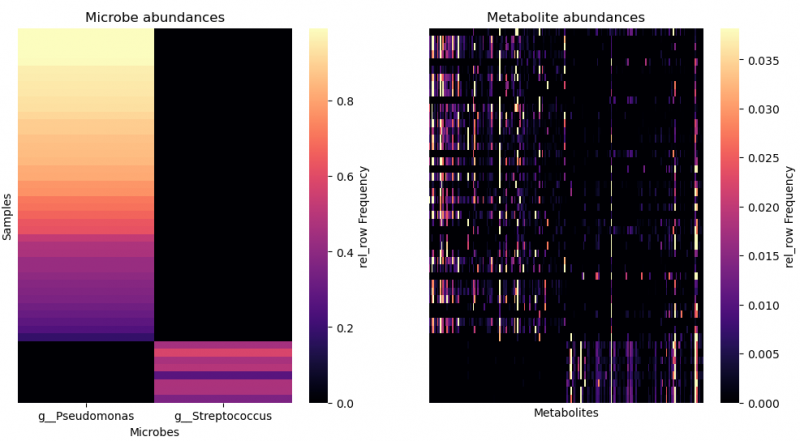
▲指定微生物及其共现代谢物 paired heatmap
2. MIMOSA 2 主要结果展示
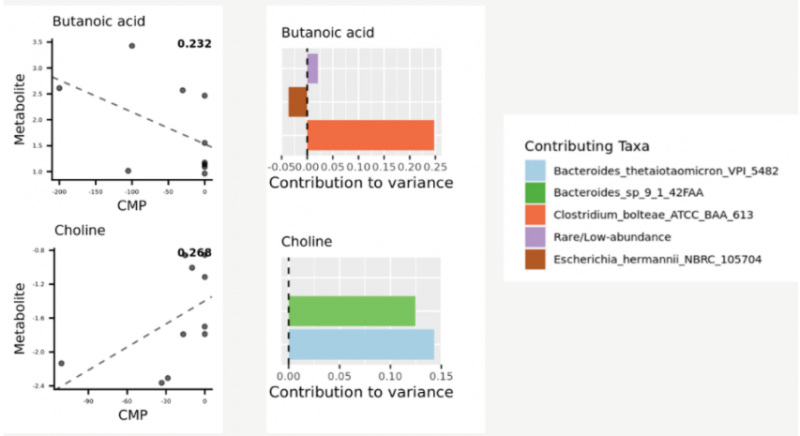
▲微生物对代谢物贡献率结果展示
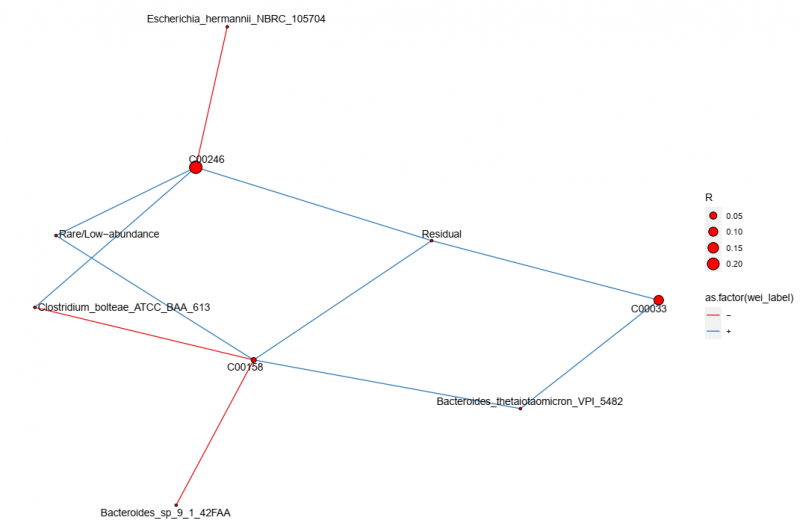
▲微生物与代谢物网络图展示
参考文献:
1. 侯璐文,吴长新,秦雪梅,等。肠道微生物功能宏基因组学与代谢组学关联分析方法研究进展 [J]. 微生物学报,2019, 059(009):1813-1822.
2. Morton J T , Aksenov A A , Nothias L F , et al. Learning representations of microbe–metabolite interactions[J]. Nature Methods, 2019, 16(12).
3. Noecker C , Eng A , Srinivasan S , et al. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation[J]. Msystems, 2016, 1(1).
更多伯豪生物人工服务:

