前面我们有介绍了利用 10x Space Ranger 软件分析空间转录组原始数据得到可用于下游分析的矩阵和镜像文件。今天来介绍一下怎么利用 Space Ranger 的结果文件进行后续分析,这里主要使用 Seurat 在进行下游分析。
先来回顾一下跑完 Space Ranger 得到哪些结果文件:
Outputs:
|
用 seurat 进行下游分析主要用到两个结果文件。一个是 filtered_feature_bc_matrix.h5 文件,一个是 spatial 镜像结果目录。
安装 R 包
由于 seurat 分析空间转录组的 R 包 satijalab-seurat 是在 GitHub 上的,如果我们需要直接安装的话需要先安装 R 包 devtools,然后利用 devtools 工具中的 install_github 来安装 GitHub 上的 R 包。
安装 devtools
install.packages('devtools')安装 satijalab-seurat
devtools::install_github("satijalab/seurat",ref = "spatial")考虑到直接安装 github 上的 R 包速度是很慢的,非常考验网速,可能需要多次才能安装成功,我们也可以直接下载安装包,本地安装。
# 下载https://codeload.github.com/satijalab/seurat/legacy.tar.gz/spatial# 安装install.packages("satijalab-seurat-v3.1.5-351-g85610bc.tar.gz", repos = NULL, type = "source")
注意该 R 包还在开发中,不要和之前安装的 seurat 包冲突。
数据准备
这里使用从 10x 官网下载的小鼠脑组织样本 MouseBrain Serial Section 1 (Sagittal-Posterior)。
下载网址:https://support.10xgenomics.com/spatial-gene-expression/datasets
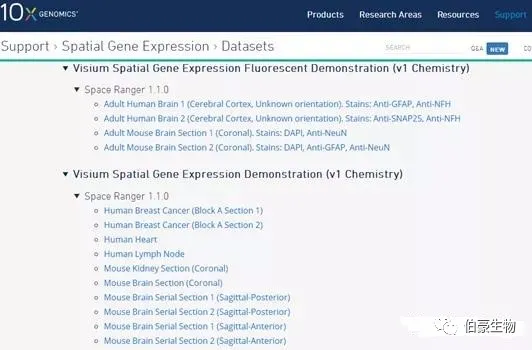
点击选择的样本,下载两个数据就行:
cell matrix HDF5 (filtered)和 Spatial imaging data
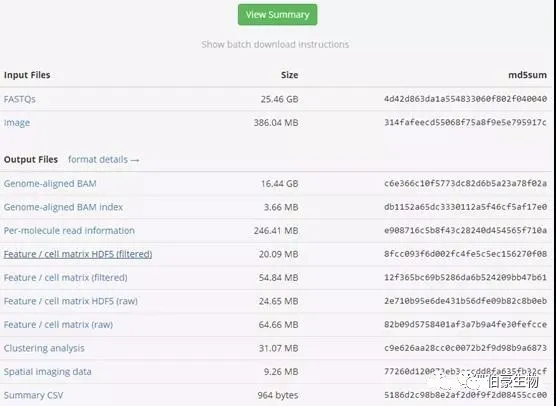
导入 R 包,读取文件
library("Seurat")library("ggplot2")library("cowplot")library("dplyr")library("hdf5r")## 读取矩阵文件name='Posterior1'expr <- "/pubj/ST_test/RNA/Sagittal-Posterior1/V1_Mouse_Brain_Sagittal_Posterior_filtered_feature_bc_matrix.h5"expr.mydata <- Seurat::Read10X_h5(filename = expr)mydata <- Seurat::CreateSeuratObject(counts = expr.mydata, project = 'Posterior1', assay = 'Spatial')mydata$slice <- 1mydata$region <- 'Posterior1' #命名# 读取镜像文件imgpath <- "/pubj/ST_test/RNA/Sagittal-Posterior1/spatial"img <- Seurat::Read10X_Image(image.dir = imgpath)Seurat::DefaultAssay(object = img) <- 'Spatial'img <- img[colnames(x = mydata)]mydata[['image']] <- imgmydata #查看数据An object of class Seurat32285 features across 3355 samples within 1 assayActive assay: Spatial (32285 features)
从 mydata 的输出信息我们可以知道,这个样本包含 3355 个 spot 点、32285 个基因。
基础统计作图
##UMI 统计画图plot1 <- VlnPlot(mydata, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
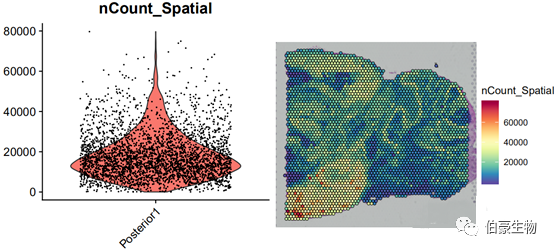
UMI 数大部分集中到 10000-20000 区间,不超过 80000,并且组织中高 UMI 数的区域主要集中在左下角。后面可以关注一下左下角区域的基因的表达和主要的细胞类型。
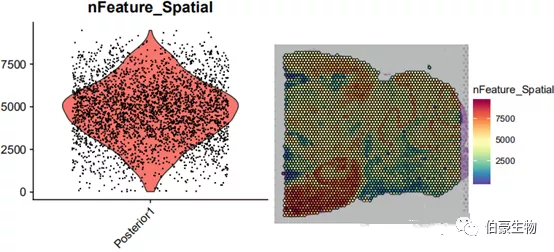
##gene 数目统计画图plot1 <- VlnPlot(mydata, features = "nFeature_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nFeature_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
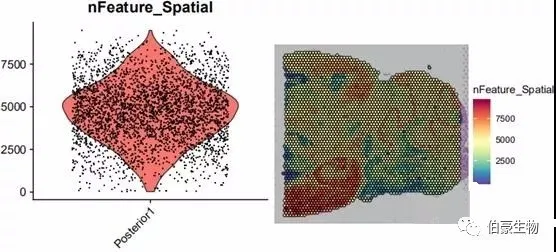
基因数目大部分处于 2500-7500 之间,结合 UMI 数据的分布可以发现 UMI 数目高的区域基因数也高,说明基因数和 UMI 数基本上是呈正相关的。
# 线粒体统计mydata[["percent.mt"]] <- PercentageFeatureSet(mydata, pattern = "^mt[-]")plot1 <- VlnPlot(mydata, features = "percent.mt", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "percent.mt") + theme(legend.position = "right")plot_grid(plot1, plot2)
注意如果是人的数据 pattern ="^mt[-] 改成 pattern ="^MT[-]
总体来说,这个样本的线粒体比例不高,左边中上区域有一处线粒体比例稍微高一点,后面也可以仔细研究一下这一块区域到底是特定的细胞类型引起的还是组织活性的差异引起的。不过从这张图我们还可以发现一个有意思的现象,基因和 UMI 高表达的区域往往线粒体比例更低。
数据过滤
做单细胞 RNAseq 我们都会根据 UMI、基因数、线粒体比例等进行过滤,那么做空间转录组数据分析其实我们也可以按这样的方式来过滤。具体的过滤条件需要根据具体样本数据来定,没有固定的标准。
比如这个样本我们可以设置过滤条件:
①基因数大于 200,小于 7500
②UMI 数大于 1000,小于 60000
③线粒体比例小于 25%
mydata2 <- subset(mydata, subset = nFeature_Spatial> 200 & nFeature_Spatial <7500 & nCount_Spatial> 1000 & nCount_Spatial <60000 & percent.mt < 25)mydata2An object of class Seurat32285 features across 2977 samples within 1 assayActive assay: Spatial (32285 features)
过滤后还剩 2977 个 spot 点。过滤后我们在绘制一下 UMI 分布图。
plot1 <- VlnPlot(mydata2, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata2, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)

那么现在问题来了,过滤之后组织图像里面缺了几块,显得特别丑。那么我们到底应不应该过滤呢?过滤数据可以减少离群的点,减少对后面聚类结果的影响,不过滤数据可以让组织图像保持完整性,绘图更好看一点,所以这个还真不好决断。
数据归一化
Seurat 官方推荐使用 sctransform 归一化方法,它构建了基因表达的正则化负二项模型,以便在保留生物差异的同时考虑技术因素。
Sctransform 函数同时实现了 NormalizeData、ScaleData、FindVariableFeatures 三个函数的功能。
mydata <- SCTransform(mydata, assay = "Spatial", verbose = FALSE)基因表达可视化
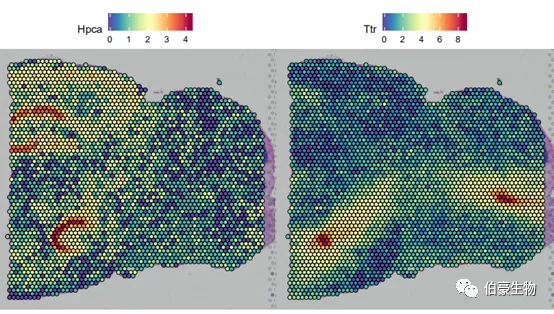
Seurat 的 SpatialFeaturePlot 功能扩展了 FeaturePlot,可以将表达数据覆盖在组织组织上。这里展示的 Hpca 基因是一个强的海马 marker,Ttr 是一个脉络丛 marker。可以通过基因的表达分布来初步判断一下海马区和脉络丛区处于组织切片的哪个位置。
SpatialFeaturePlot(mydata, features = c("Hpca", "Ttr"))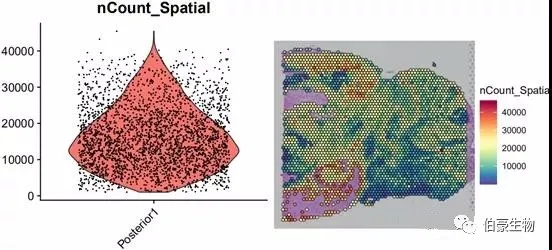
从结果的展示来看,这两个 marker 基因的分布还是挺集中的,这也说明理由空间转录组数据来分析小鼠脑的不同区域的表达差异应该还是比较准确的。另外,海马区的分布可以大概分成 3 大块,从上之下首块弧形区域似乎处于线粒体高表达区域,而下面一块弧形区处于基因高表达区。后面可以把这三个不同区域的数据进行差异基因和功能的比较也许会发现一些有意思的东西。
降维、聚类和可视化
接下来利用 seurat 进行降维和聚类。先进行 PCA 降维,再选择前 30 个维度进行聚类和 umap、tsne 降维。
mydata <- RunPCA(mydata, assay = "SCT", verbose = FALSE)mydata <- FindNeighbors(mydata, reduction = "pca", dims = 1:30)mydata <- FindClusters(mydata, verbose = FALSE)mydata <- RunUMAP(mydata, reduction = "pca", dims = 1:30)mydata <- RunTSNE(mydata, reduction = "pca",dims = 1:30)
tsne 展示结果:
Umap 展示结果:
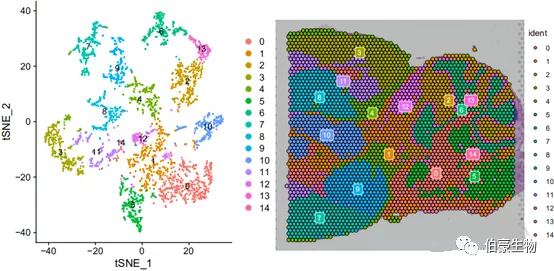
tsne 和 umap 两种展示方式在这次分析里差别不是特别大,tsne 相对来说亚群与亚群之间分的更开,而 umap 则单个亚群位置更集中。这个时候我们也可以结合前面 marker 基因的表达分布图来大概判断一下每个亚群大概处于小鼠脑的那个区。
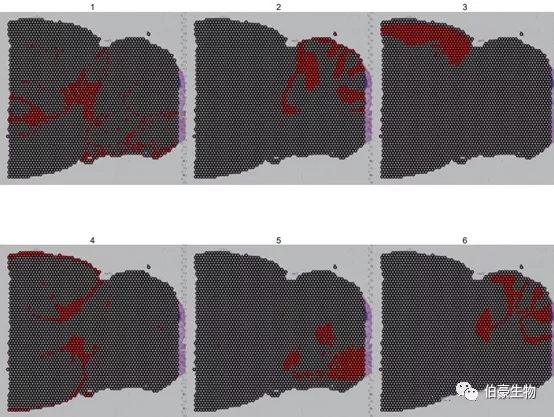
由于亚群的颜色比较接近,有时候不太好判断,我们可以是 cells.highlight 来标记特定的亚群。
SpatialDimPlot(mydata, cells.highlight = CellsByIdentities(object = mydata, idents = c(1, 2, 3, 4,5, 6)), facet.highlight = TRUE, ncol = 3)
今天先分享到这,下期继续!
更多伯豪生物人工服务:

