随着测序技术的飞速发展,单细胞转录组测序技术也已成为实验室常规工具之一。然而,研究人员在试图应用单细胞转录组技术的时候也面临着令人困惑的选择,比如说选择哪种建库测序平台,使用哪种分析方法以及后续的生物信息学分析方法的选择等等。
此前,来自人类细胞图谱联盟的研究人员进行了一项综合性多中心研究,通过使用包含人类、小鼠和狗细胞的参考样本,比较了 13 种单细胞转录组测序流程的异同。结果发现不同流程在量化基因表达和识别细胞类型层面存在着显著差异。
近日,美国罗马琳达大学基因组学中心的研究团队在 Nature Biotechnology 发表了题为“A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples”的研究性文章,研究人员设计了一项综合性的多中心研究,用以评估技术平台、样品组成和生物信息学方法(包括预处理、归一化和批次效应校正)的影响,并在后为科研人员解决科学问题的技术平台和生物信息方法的结合提供了实践指导。

文章发表在 Nature Biotechnology
该研究使用了四种测序平台:10x Genomics,Fluidigm C1, Fluidigm C1 HT 和 Takara Bio ICELL8;测序工作分别由四个研究中心完成:Loma Linda University(LLU),the National Cancer Institute(NCI),the US Food and Drug Administration(FDA)和 Takara BioUSA(TBU)。样本层面,他们使用了有两个特征明显的参考细胞系:来自同一供者的乳腺癌细胞系(样本 A)和“正常”B 淋巴细胞系(样本 B)。然后使用 3 '或全长单细胞转录组测序方法对 30,693 个单细胞进行了测序,共生成了 20 个数据集。
针对产生的这 20 个数据集,研究人员对不同的数据预处理方法、数据标准化方法、批次效应矫正方法等进行了评估。
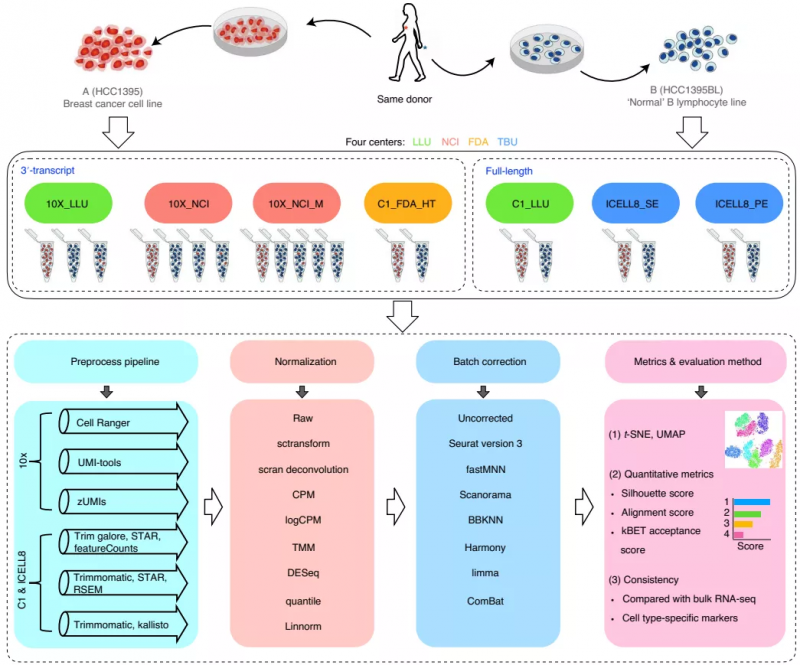
图 1. 研究总体设计示意图。来源:Nature Biotechnology
测序深度与检测基因数的关系
首先,研究人员对序列深度与检测到的基因数量的关系进行了评估。正如预期的那样,随着测序深度的增加,检测到的基因数逐渐升高并趋于稳定。另外,对于癌细胞(样本 A)和 B 淋巴细胞(样本 B),随着测序深度的增加,每个细胞检测到的基因数量迅速增加,特别是 Fluidigm C1 平台。然而,对于全长测序技术(C1_LLU 和 ICELL8),在 10 万次读取后,饱和速率较慢,在相同的测序深度增加情况下,与基于 3’的测序技术相比,额外能够检测到的基因较少。
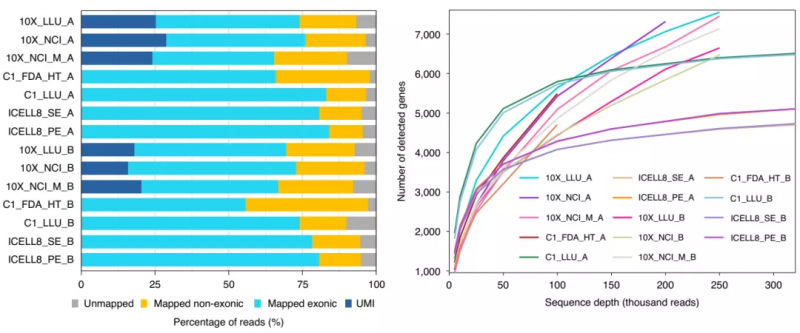
图 2. 不同测序平台检测的基因数及与测序深度的关系。来源:Nature Biotechnology
数据预处理方法的比较
对基于 UMI(Unique Molecular Identifier)的单细胞转录组测序数据,他们比较了三种预处理方法:Cell Ranger 3.1(10x Genomics)、UMI-tools 和 zUMIs。结果显示,三种方法在识别细胞数量和每个细胞检测到的基因数量层面都存在差异。不过,Cell Ranger V3 是灵敏的细胞条形码识别方法,UMI-tools 和 zUMIs 可以过滤大多数低基因或转录表达的细胞,但每个细胞内可检测到更多的基因。
对非基于 UMI 的单细胞转录组测序数据,他们比较了另外三种预处理方法:featureCounts、kallisto 和 RSEM。这些数据预处理流程包括去除低质量测序数据、基因组比对和基因计数。结果表明,三个不同的预处理方法检测到的基因数量的差异比较大。kallisto 在全长转录组测序数据中发现了每个细胞中更多的基因。此外,基于 Fluidigm C1 HT 3’测序方法产生的数据中,kallisto 方法检测到的每个细胞的基因数与其它两个管道生成的基因序列有显著差异。
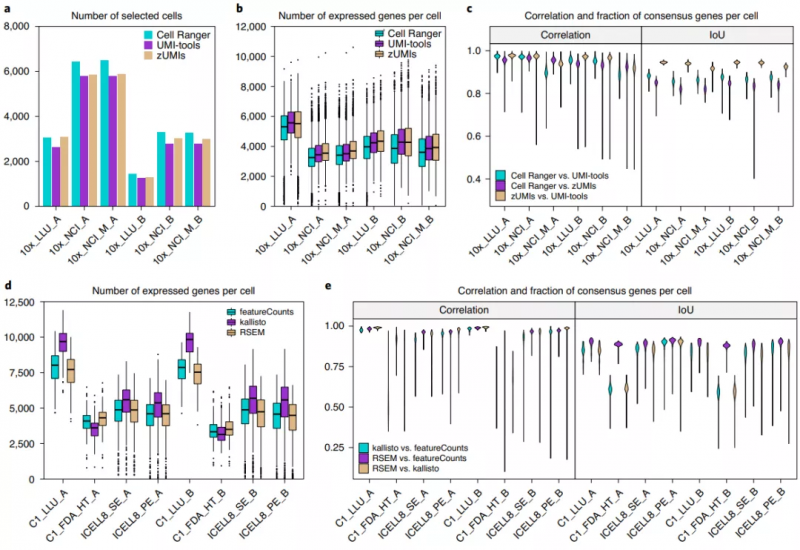
图 3. 数据预处理方式对检测到的基因数量的影响。来源:Nature Biotechnology
不同批次矫正算法的比较
如上所述,数据集之间的差异可能来自技术层面或生物因素,针对这些技术层面带来的差异,在进行数据分析时是需要矫正的,否则将会影响结论。研究者对七种校正批次效应的算法进行基准测试:Seurat version 3、fastMNN、mutual nearest neighbors(MNN)、Scanorama、BBKNN、Harmony、limma 和 ComBat。
他们通过四种不同的样本组合评估这些算法的性能,组合 1 包含所有单细胞转录组数据集,包括混合和纯合数据集;组合 2 只包含了乳腺癌细胞系数据;组合 3 分别对 B 细胞系来源数据进行评估;组合 4 中,数据由将 5% 或 10% 的乳腺癌细胞(样本 A)加入到 B 淋巴细胞(样本 B)中,用 10x Genomics 平台横跨两个中心测序得到。
结果显示,在去除批次效应和从 B 淋巴细胞中分离乳腺癌细胞方面,BBKNN、fastMNN 和 Harmony 是有效的;Seurat V3 是将不同批次的相似细胞聚集在一起的方法之一,特别是对乳腺癌细胞,但也会存在过度校正的现象,比如将两种高度不同的细胞类型融合在一起。另外,当只分析来自 10x 平台的数据时,Scanorama 既能清晰地分离不同的细胞,又能很好地将相似的细胞组合在一起。
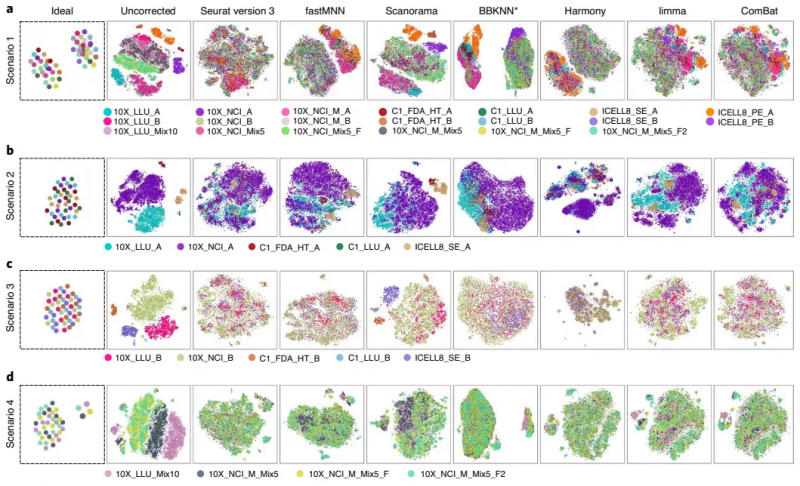
图 4. 比较分析不同工具的批次矫正效果。来源:Nature Biotechnology
综合上述的分析结果,研究人员对这些预处理方法和算法进行了综合排序,如图 5 所示,基于 UMI 的数据可以用文中所列的任何方法进行预处理,而 kallisto 则更适用于全长转录组测序数据的预处理。
在跨中心数据集,特别是当数据集中存在大量不相似细胞时,BBKNN 表现先进,而 limma 和 ComBat 在两种类型的细胞的跨平台、跨中心分离中表现差。Seurat V3、fastMNN 和 Harmony 都能很好地混合来自不同平台和位点的生物相同或相似样本的单细胞转录组数据。
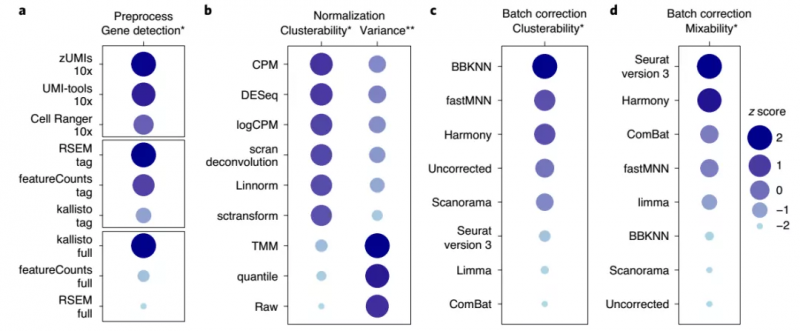
图 5. 生物信息学指标的性能排名。来源:Nature Biotechnology
综上所述,该研究比较分析了 6 种单细胞转录组数据预处理流程、8 种归一化方法和 7 种批次校正算法,结果表明,单细胞转录组数据之间的确存在批次效应,不过,跨中心和不同平台的数据差异可以通过适当的计算方法进行纠正。同时,该研究也强调了选择适合的测序技术平台和分析数据算法的重要性。如下图所示,他们也根据本研究结果为科研人员选择适合解决科学问题的技术平台和生物信息方法的结合提供了实践指导。
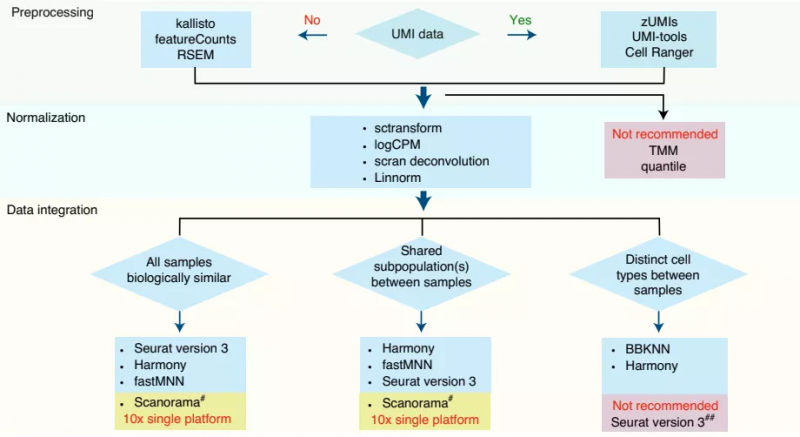
图 6. 好的分析推荐方案。来源:Nature Biotechnology
参考文献:
1.Chen, W., Zhao, Y., Chen, X. et al. A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nat Biotechnol (2020).
2.Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch efects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
3.Butler, A., Hofman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across diferent conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
转载:测序中国(侵删)

更多伯豪生物人工服务:

