空间转录组技术的出现,使研究人员了解不同转录本的空间位置信息。但是,目前的技术还达不到单细胞的分辨率水平。也就是说,探针在特定位置捕获到的转录组是来源于多个细胞的,而且这些细胞可能来自一组异质性的细胞,而并非同一类型的。因此,在组织任何位检测到的基因表达谱都可以被认为是来自多个不同细胞(相同类型细胞或不同类型细胞)来源的转录本的混合物。这也就意味着,即使转录产物的空间位置信息可以完全绘制成图表,但是产生这种转录的细胞的类型和空间分布仍然很大程度上是未知的。
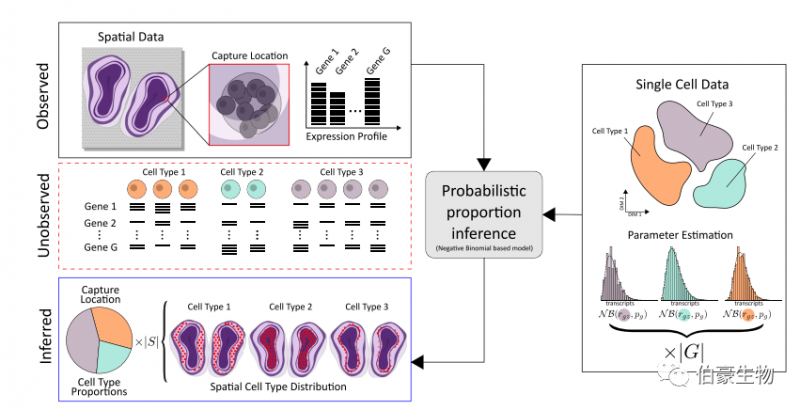
如上所述,空间转录组学技术面临着一个困境,即知道转录本的位置,但不知道是哪个细胞产生了它们。相反,单细胞 RNA 测序可以将每个转录本与单个细胞联系起来,但是关于这些转录本在组织中的位置的信息丢失了。鉴于这种互补的优势和劣势,结合两种技术的数据来描绘细胞类型群体的空间信息是很好的选择。
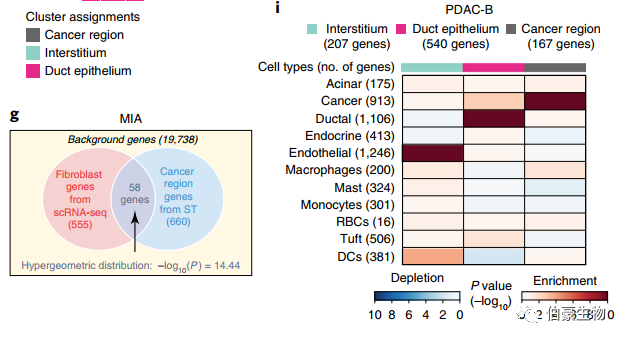
先前研究已经报道了,基于 bulk RNA-seq 数据来鉴定细胞类型的方法。发表在 nature biotechnology 上的一篇关于胰腺导管癌的单细胞和空间转录组的研究,基于 MIA(Multimodal intersection analysis)的算法也可以将细胞类型与空间位置对应起来。这种分析首先通过描述细胞类型特异性基因和组织区域特异性基因,然后确定它们的重叠是比偶然预期的高(富集)还是低(缺失)。在 scRNAseq 数据中,定义基因集的方法是,在每个细胞类型中,在标注到该细胞类型的细胞中,与其他细胞的表达量相比,其表达量在统计学上更高(P<10e−5)。对于 ST 数据,确定了每个空间区域相对于其他区域表达显著更高的基因(P<0.01,双尾 student t 检验)。通过 scRNA-seq 和 ST 提取的基因集,MIA 接下来计算每对细胞类型特异性和区域特异性基因集之间的重叠,并执行超几何测试来评估显著富集或缺失。[1]
发表在 COMMUNICATIONS BIOLOGY 杂志上的一篇文章提出了一种新的替代的基于模型的方法来整合单细胞 RNA 测序和空间转录组测序数据,它利用完整的表达谱而不是精选的一组标记基因。使用这种方法,研究人员对来自小鼠大脑和发育心脏的单细胞数据中的细胞类型空间映射到相应的组织切片上。并证实该方法优于其他方法 [2]。
在未来,单细胞测序联合空间转录组测序会是全面解析样本中细胞类型和空间位置信息的新方法,也是突破已知,探索未知的新技术领域。
参考文献
【1】Moncada, R., Barkley, D., Wagner, F. et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol (2020) doi:10.1038/s41587-019-0392-8
【2】Alma Andersson1, Joseph Bergenstråhle1 et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. COMMUNICATIONS BIOLOGY (2020) doi.org/10.1038/s42003-020-01247-y
伯豪生物 - 空间转录组测序服务优势
高质量切片: 切片、贴片经验丰富,针对不同组织优化了解决方案。
流程化分析: 完善的分析流程,准确快速解析空间转录组数据。
标准化内控: 丰富的实操经验构建了标准化的内控体系。
专业化团队: 资深的技术团队具有多年项目方案设计、实验操作、售后分析等经验。
全流程服务: 提供组织冷冻包埋、贴片、切片、透化、建库测序及数据分析的全套服务。
更多伯豪生物人工服务:

