 |  |

伯豪生物官网 www.shbio.com 单细胞 ATAC 测序技术服务来了!
单细胞 ATAC 测序(Aaasay for transposase Accessible Chromatin with high throughput sequencing at the single cell level)翻译成中文是在单细胞水平上通过高通量测序技术来研究染色质开放程度(也叫染色质的可及性)。染色质开放程度(染色质的可及性),反映了染色质的转录活性状态,是研究基因表达调控的重要方向,在表观遗传图谱绘制、细胞分化和发育及各类疾病的发生发展研究中具有重要的作用。
对染色质可及性的研究是伴随着对染色体结构研究的发展逐渐兴起的。1971 年,Mirsky 首先使用 DNase 来研究染色质的结构,发现 DNase 对于存在与染色中的 DNA 仍然可以切割,表现出染色中的 DNA 对于 DNase 的可及性。1975 年,Burkholder 和 Weaver 研究发现 DNase I 对舒展状态的染色质的消化速率高于对压缩状态的染色质。同时指出 DNA 与染色质蛋白的结合程度的差异与这两种状态下染色质片段的功能相关。目前人们已经知道双螺旋的 DNA 与组蛋白结合后,会以染色质或染色体的形式形成高级空间结构。以人的基因组为例,每个组蛋白八聚体上缠绕有 146 个碱基对的 DNA。连接核小体与核小体之间的 DNA 序列称为连接序列。活细胞中染色质的结构总是处在动态变化中,在不同类型的细胞中,或在不同的生理条件和外界刺激下,细胞核中染色中呈现不同的结构和状态。这些结构和动态变化的状态表现形式之一就是染色质可及性的变化。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
2015 年 4 月,Science 发表了 Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing [1] 的文章。同年 7 月,Nature 发表了 Single-cell chromatin accessibility reveals principles of regulatory variation [2] 的文章。这两篇论文先后提出利用单细胞 ATAC-seq 技术对染色质可及性进行检测,探索细胞转录调控机制,解决了以往存在的细胞异质性难题,成为 ATAC-seq 技术的一大突破。其中,后者将 ATAC-seq 与 Fluidigm C1 单细胞平台整合,利用微流控芯片完成捕获、裂解、转座、PCR 等实验过程,建立了自动化的单细胞染色质可及性图谱研究方法。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
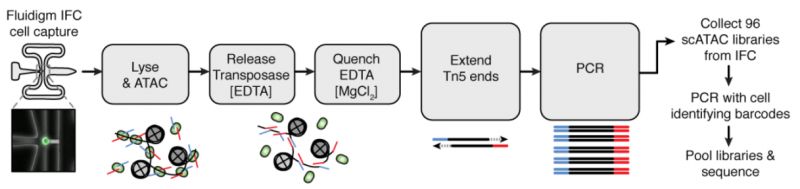
图 ATAC-seq 与 Fluidigm C1 单细胞平台整合的实验流程 [2]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
作者首先对 254 个类淋巴母细胞进行了单细胞 ATAC 测序,将这些单细胞数据合并分析后得到的结果与群体细胞 DNase-seq 或 ATAC-seq 获得的染色质可及性图谱具有很高的相关性,单细胞水平的数据再现了一些群体细胞 ATAC-seq 数据反映出的染色质特征。
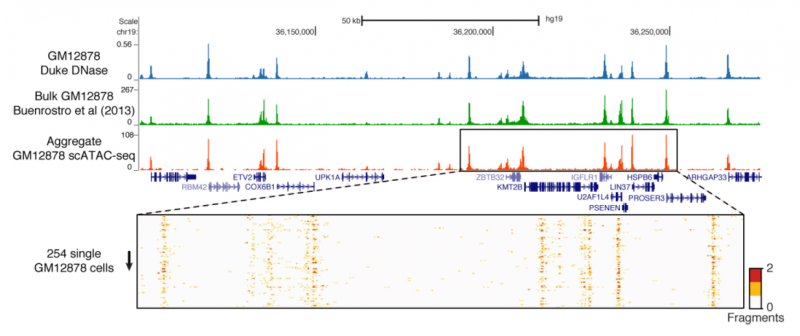
图 单细胞 ATAC-seq 与常规 ATAC-seq 的一致性 [2]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
为了进一步验证方法的可靠性,作者又用 scATAC-seq 的方法对 ENCODE 细胞系,包括 H1 人类胚胎干细胞、K562 慢性粒细胞性白血病细胞、GM12878 类淋巴母细胞、V6.5 小鼠胚胎干细胞、EML1 细胞(小鼠造血祖细胞)、TF- 1 细胞(人类成红细胞)、HL-60 cells(人类 promyeloblasts)和 BJ 成纤维细胞 HL-60 细胞进行了分析。结果发现在增殖细胞中,复制时序结构域(replication timing domains)的染色质可及性的变异性增加。同时,作者还发现不同的转录因子可以通过协同或者竞争性结合的作用促进或者抑制染色质可及性的可变性。通过此方法对作者对大量转录因子的 ChIP-seq 数据研究绘制出了转录因子协同作用改变染色质可接近性的图谱。此外,还发现与高可变性相关的转录因子的是细胞类型特异的,在单细胞中染色质状态与组蛋白修饰也与染色质可接近性变化相关。
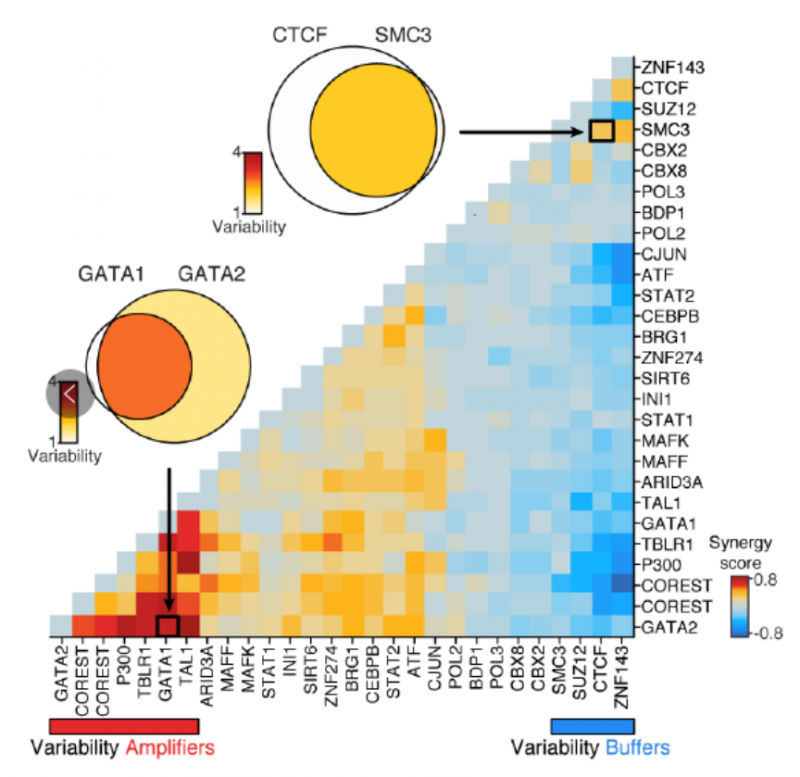
图 转录因子通过协同或者竞争性结合作用促进或者抑制染色质可及性的可变性 [2]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

2018 年 10 月,10X Genomics 推出的 Chromium 单细胞 ATAC 解决方案提供了一种全面的、可扩展的方法来研究分析单个样本中成百上千个细胞中染色质的开放情况。通过转座酶对混合的细胞核悬液进行核 DNA 的切割,然后使用微流控芯片将溶液中的细胞核包裹到油滴中,形成纳米级的凝胶珠状乳状液(GEMs)。采用 10X 条形码,对每个细胞核切割的 DNA 加上条形码。通过文库构建,测序,根据 10X 条形码将测序得到的序列关联到每个单独的细胞核上。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

1、转座酶切割核 DNA
细胞核悬浮液在包括转座酶的混合液中孵育。转座酶进入细胞核,优先在染色质的开放区域将 DNA 片段化。同时,将测序接头序列添加到片段化的 DNA 片段的末端。
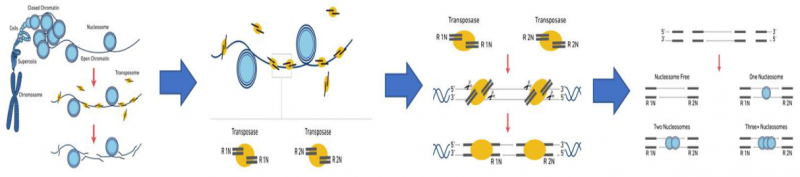
图 转座酶切割 DNA 示意图
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
2、GEM 生成及 Barcode 添加
在 Chromium Next GEM Chip H 芯片上,使包含有 barcode 的凝胶珠,转座酶切割后的细胞核,混合液(包括 ATAC buffer 和 ATAC 酶),以及油滴进行混合,生成 GEMs。为了达到每个油滴中包含有单个细胞核,需要对细胞核进行有限稀释,保证生成的大多数 GEMs(~90-99%) 不包含细胞核,而其余的为包含单个细胞核 GEMs。

伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
GEMs 生成后,凝胶珠会溶解释放出含有(i) Illumina P5 序列、(ii) 16nt 10X 条形码序列和(iii) Read 1 (Read 1N) 序列的寡核苷酸。这些核苷酸序列会于片段化的 DNA,以及混合液进行混合。后续经过热循环后生成含有 10X barcode 的单链 DNA。经过孵育后,对 GEMs 进行破油处理,所有 GEMs 中的含有 10X barcode 的单链 DNA 混合在一起,并进行回收。
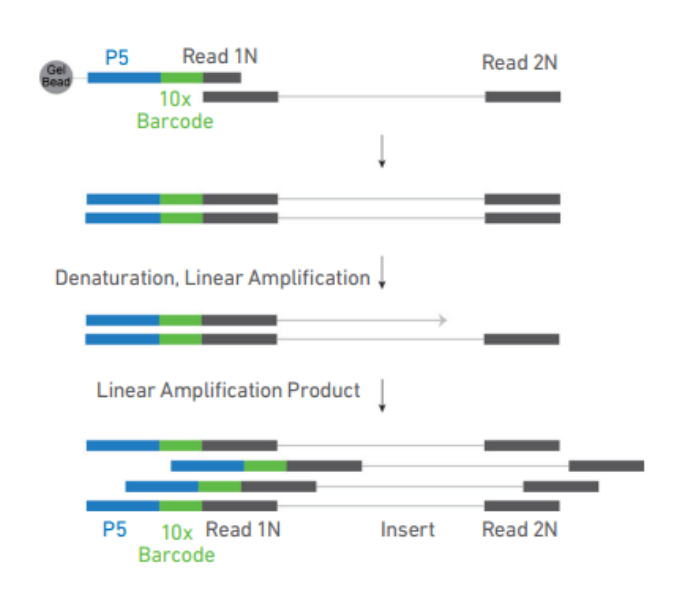
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
3、破油后的纯化
使用硅烷磁珠清除破油反应混合物中残留的生化试剂。固相可逆固定(SPRI) 珠子用于从样本中消除未使用的 10X 条形码。
4、测序文库的构建及质检
通过 PCR 将 P7 接头以及样本的标签(Index N)添加到文库的两端,形成包含有 P5 和 P7 接头序列的文库,用于 Illumina 桥式 PCR 扩增。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
文库结构
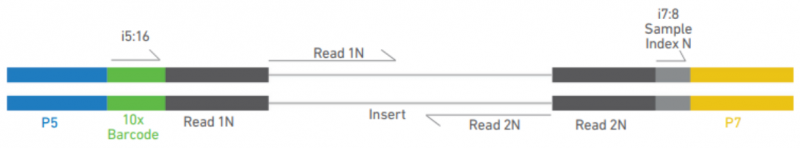
图 ATAC 文库组成示意图
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
通过 Agilent TapeStation High Sensitivity D1000 ScreenTape 对文库进行质检,结果如下:
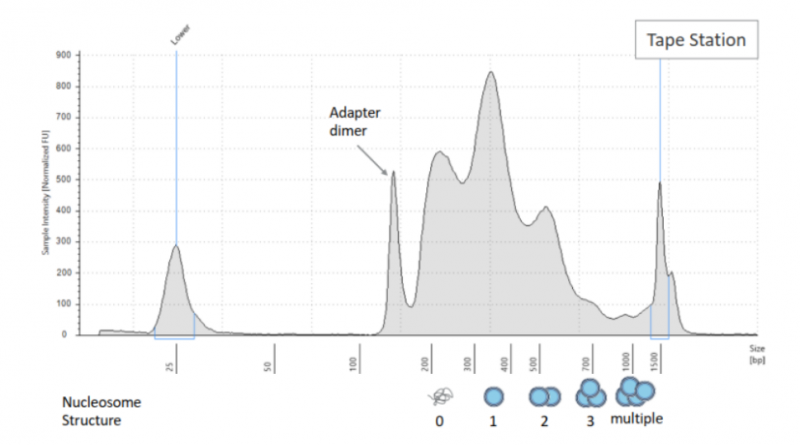
图 Agilent TapeStation High Sensitivity D1000 ScreenTape 文库质检结果
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
或者用 Agilent Bioanalyzer High Sensitivity DNA chip 来检测片段大小,结果如下:
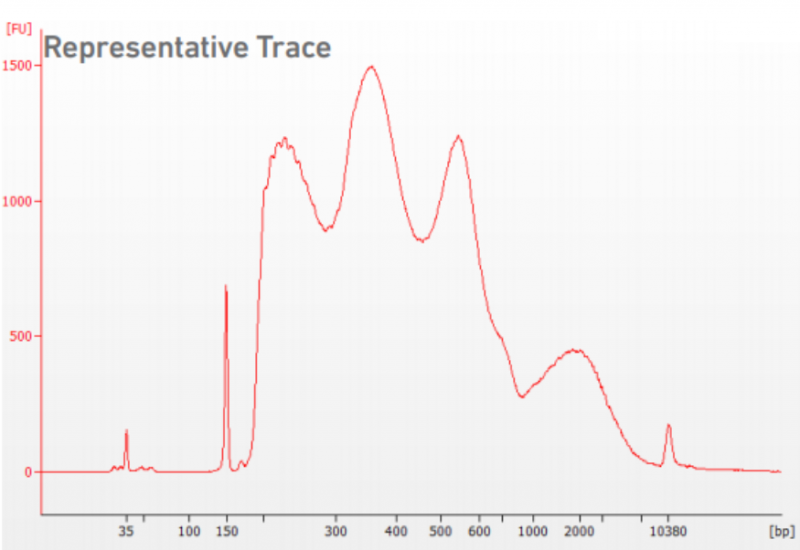
图 Agilent Bioanalyzer High Sensitivity DNA chip 质检结果
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
备注:
a、横坐标表示文库的片段程度,其中 0 代表核小体 free 的峰。1 代表包含有一个核小体的峰;2 代表包含有 2 个核小体的峰;以此类推。
b、核小体是由 DNA 和组蛋白形成的染色质基本结构单位。每个核小体由 146bp 的 DNA 缠绕组蛋白八聚体 1.75 圈形成。核小体核心颗粒之间通过 50bp 左右的连接 DNA 相连。加上两端的 P5,P7 接头,barcode,sample index,R1N 序列,长度大概如下:核小体 free 的峰长度 200 多 bp;1 个核小体的峰约为 300 多 bp;2 个核小体的峰约为 500bp,以此类推。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
建议测序深度及参数
指标 |
参数 |
测序深度 |
每个细胞核测 25000 read pairs |
测序类型 |
PE150,dual-index(双 index 测序) |
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

▲流程精简时效快: 可检测单细胞转录调控区域中的开放性染色质。
▲通量高: 每个通道 500-10000 个细胞核。
▲效率高: 细胞核捕获率高达 65%。
▲适用范围广: 经验证适用于原代细胞,冻存细胞,新鲜组织等。
▲信息分析: 获取信息量大,可精细化分析。
▲同一份样本可实现单细胞 ATAC、mRNA、TCR/BCR 同时测序,并整合数据。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

▲类型: 新鲜组织,原代细胞,细胞系等。
▲来源: 血液提取、磁珠富集、流式富集、组织解离等。
▲样本量及其它质控要求:
| 样本类型 | 样本质控要求 |
| 细胞悬液 | >1x105 个细胞; |
| 活率 >80%; | |
| 浓度 500~1,000 个细胞 /ul; | |
| 细胞间无粘连(成团率 <5%); | |
| 无大于 40um 的细胞碎片或其他颗粒物; | |
| 不存在逆转录抑制剂和非细胞的核酸分子。 | |
| 血液 | EDTA 抗凝的全血(不可肝素抗凝),>5ml。 |
| 组织 | 0.3cm×0.3cm(不超过 0.5cm×0.5cm)的新鲜组织,4~5 块。 |
▲样本的保存与运输:
(1)细胞悬液:建议现场制备,如要运输,建议使用伯豪生物自主研发的单细胞保护液,4°C 运输,48 小时内送达伯豪生物实验室。
(2)血液:EDTA 抗凝的全血,4°C 运输,2 小时内送达伯豪生物实验室;或提取 PBMC 后冻存,干冰运输。
(3)组织:建议使用伯豪生物自主研发的单细胞 ATAC-seq 组织保护液,4°C 运输,48 小时内送达伯豪生物实验室。
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

▲干细胞 / 发育生物学
▲肿瘤学
▲免疫学
▲神经科学
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

1、The Cell Ranger ATAC 分析结果
10X genomics 官方的 Cell Ranger ATAC 流程会输出一个 HTML 文件,其中包含数据统计结果和初步的分析结果。
a、基本数据统计(细胞数,每个细胞测到的数据的中位值,比对到 peaks 上的数据的比例等)
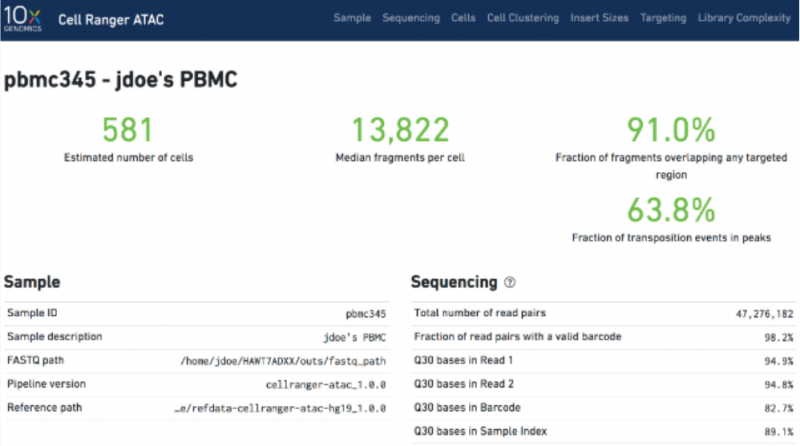
图 单细胞 ATAC 基本数据展示
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
b、插入片段长度统计(包括核小体 free 的比例,单核小体的比例)
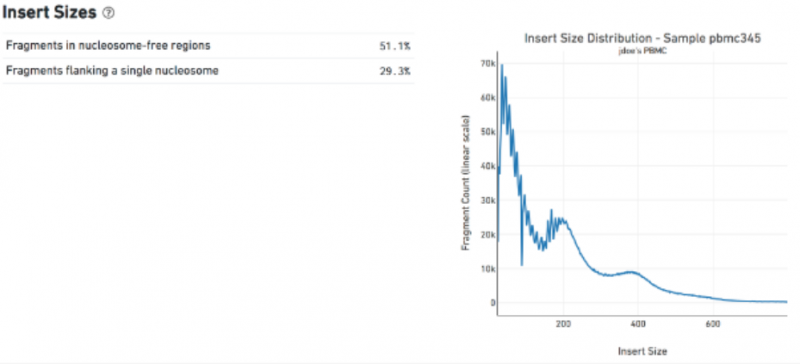
图 插入片段长度统计(单细胞 ATAC 测序数据分析)
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
c、细胞聚类结果
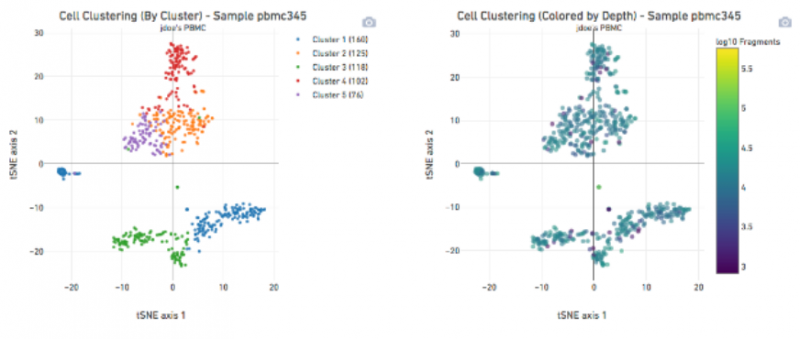
图 细胞聚类结果(单细胞 ATAC 测序数据分析)
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
2、Loupe Cell Browser 展示结果
Loupe Cell Browser 是一个适用于 Windows 和 MacOS 的桌面应用程序,它可以快速、轻松地可视化和分析 10X Chromium 单细胞 ATAC 数据。它在寻找显著的峰和识别转录因子基序、识别细胞类型、比较各组细胞之间的染色质可及性以及探索细胞簇内的子结构方面进行了优化。
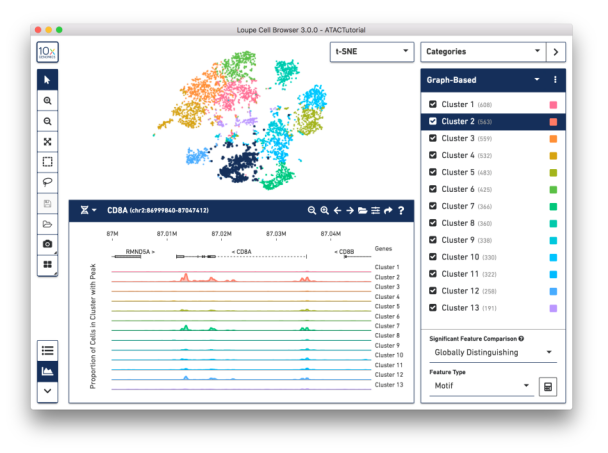
图 Loupe Cell Browser 展示细胞分群及 peaks
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
基于 RNA 的细胞注释结果,对 ATAC 的细胞类型进行打分注释。
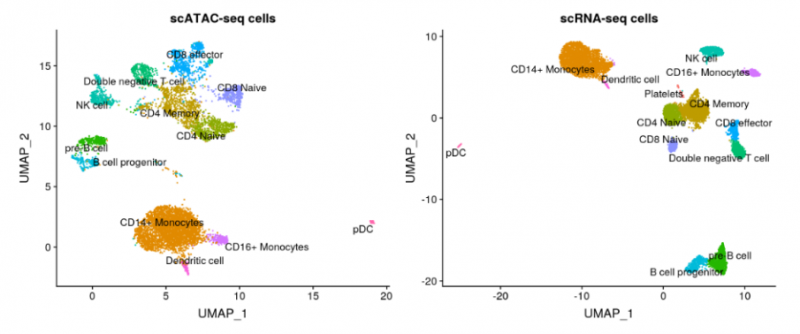
图 单细胞 ATAC 测序细胞类群注释结果
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

1、干细胞分化
造血分化是从造血干细胞分化为具有不同功能的细胞过程。造血分化过程是一个复杂、多阶段的,受多种因子调控的过程,单细胞组学技术有助于解析造血干细胞转录和细胞命运异质性的顺式和反式调节机制。2018 年,斯坦福大学的研究团队从健康人捐赠的骨髓中分选单个细胞进行 scATAC-seq[3],获得了造血系统 10 个细胞类型的染色质可及性图谱,构建了人类造血染色质可及性景观图来表征分化轨迹。作者利用 ChromVAR 鉴定不同细胞的 TF motif,发现了造血分化中主要的调控因子 GATA1, BATF, CEBPB 等。采用多种聚类方法对数据进行降维聚类分析,观察到髓系共同祖细胞(commonmyeloid progenitors,CMP) 和粒细 - 巨噬细胞祖细胞(granulocyte-macrophage progenitors,GMPs) 的异质性,并绘制了各个谱系细胞分化连续轨迹。此外本研究整合了 scATAC-seq 和 scRNAseq 数据,将髓系分化基因的动态表达映射到染色质的动态变化,并且发现了已知的髓系分化调节因子的表达模式。将转录因子表达与转录因子 motif 对比,共发现了 14,005 个顺式调控元件,这些调控元件随着染色质开放状态的变化也呈现显著的异质性。总的来说,这项工作为在单细胞分辨率下对人类原代组织复杂的调节动力学进行综合研究提供了一个框架。
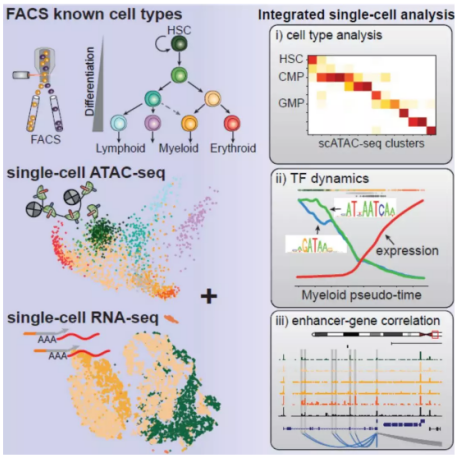
图 联合 scATAC-seq 与 scRNA-seq 研究造血分化 [3]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
2、肿瘤异质性
乳腺癌具有高度异质性,至少可分为六种不同的固有亚型,即 luminal A、luminal B、HER2-enriched、basal-like、normal breast 和 claudin-low。乳腺癌起源于乳腺上皮,人类和小鼠中的乳腺上皮,由两个主要的细胞分层构成导管上皮网络,分别是内层管腔细胞和外层基底 / 肌上皮细胞。一系列的研究表明,在小鼠的这两个细胞层中存在进一步的异质性。本研究应用单细胞转录组测序(scRNA-seq)和单细胞染色质可及性测序(scATAC-seq)对分离的乳腺上皮细胞(mammary epithelial cell,MECs)进行分析 [4],重建了小鼠 MEC 系统的细胞类型及其潜在的基因调控特征。并且在管腔细胞的分泌类型中定义了新的分化状态,将管腔细胞分为祖细胞和成熟分泌细胞簇。通过整合 scRNA-seq 和 ATAC-seq,确定了在特定上皮细胞类型以及新定义的管腔分化状态中差异激活的 cis 和 trans 调节元件。这项工作提供了一个重要资源来揭示与 MEC 身份和分化相关的调节元件,这将为确定乳腺癌中染色质可及性的变化提供有价值的参考。
![图 scATAC-seq 与 scRNA-seq 整合分析 [4]](/storage/image/2020/05/oewiUUQ0eIKFdRr2p06GHk6SO33WJyRHszxpgHUF.png)
图 scATAC-seq 与 scRNA-seq 整合分析 [4]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
3、免疫学
近期美国斯坦福大学的研究团队利用 10x Genomics 单细胞 ATAC-seq 技术,绘制了来自血液、基底细胞癌组织的 200,000 多个单细胞的染色质可及性图谱 [5]。文章对来源于 16 个健康人外周血及骨髓细胞的 63,882 个细胞核样本进行单细胞 ATAC 测序分析,基于染色质开放程度,鉴定出 31 个细胞亚群,并深入探索了免疫细胞谱系的调控轨迹。此外,研究团队还对 PD- 1 治疗前后采集的基底细胞癌患者的原发性肿瘤样本进行了检测,通过分析 37,818 个细胞的 ATAC-seq 数据,发现来源于不同患者的基质细胞、免疫细胞基本聚到一起,而肿瘤细胞的分群则表现出显著的异质性。在 PD- 1 免疫治疗后,对治疗有相应的患者中出现两种 T 细胞亚群(耗竭性 CD8+ T 细胞和 CD4+ 滤泡辅助 T 细胞)的扩张,且比例相当,暗示这两种细胞类型在 PD- 1 阻断后,其分化过程可能处于一致的调控模式。
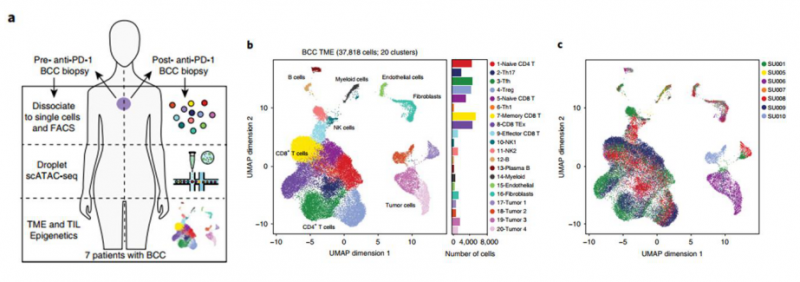
图 肿瘤细胞的的异质性及 PD- 1 治疗前后细胞亚群的改变 [5]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。
4、神经科学
利用单细胞 ATAC-seq 技术对成年雄性小鼠 13 个组织的单细胞染色质可及性进行了分析 [6]。结果共鉴定出 85 个亚群和 40 万种调控元件。将单细胞染色质可及性与单细胞转录组比较分析,发现两种方法注释的细胞类型表现出高度一致性。为了研究神经元细胞中染色质可及性的异质性,对前额叶皮质细胞的数据进行分析,结果发现,兴奋性神经元和中间神经元明显地与神经胶质细胞、小胶质细胞和内皮细胞分离。并且在兴奋性神经元内仍存在显著的异质性,可能反映了前额叶皮质不同层中的表达和甲基化差异。

图 前额叶皮质细胞中染色质可及性的异质性 [6]
伯豪生物提供:单细胞 ATAC 测序、生信分析全程技术服务,承接批量科研、临床样本,伯豪生物单细胞 ATAC 测序技术服务优质供应商。

[1] Cusanovich DA, Daza R, Adey A, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348(6237):910-4.
[2] Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523(7561):486-90.
[3] Buenrostro JD, Corces MR, Lareau CA, et al. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 2018, 173(6):1535-1548.
[4] Nicholas P, Quy H, Guadalupe G, et al. Integrated single-cell transcriptomics and chromatin accessibility analysis reveals novel regulators of mammary epithelial cell identity. Biorxiv 2019 Aug 20; preprint. biorxiv: 10.1101/740746v1.
[5] Satpathy AT, Granja JM, Yost KE, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 2019, 37(8):925-936.
[6] Cusanovich DA, Hill AJ, Aghamirzaie D, et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility.Cell 2019, 174:1309-1324.
-END-

